Overview
An understanding of factors that can impact product purity, is a crucial component of assuring the quality of pharmaceuticals released to market. Regulatory bodies such as the US FDA and the European Medicines Agency (EMA) are increasingly focusing on the interactions between the various steps involved in manufacturing the final product, to assess whether or not companies have a thorough understanding of the toxicological risks that can occur from Extractables and Leachables.

Regulators and international bodies are routinely publishing new/revised regulations and guidance documents, and in particular for the biopharmaceuticals industry, due to the complex nature of the manufacturing process and the protein (drug product). The focus is now not just on the final container (primary packaging) but also the effect of the process stream, particularly with biologics and the increased use of single use systems/technologies.
This article will give an overview of Extractables and Leachables focusing on the biopharmaceutical industry.
Let’s look at the definitions of Extractables and Leachables (let’s call them E/L) to clear up any confusion as sometimes they are referred to as the same thing - which they are not! Regulatory bodies in general define E/L in the following way:
Extractables – Organic and inorganic chemical entities that are released from a pharmaceutical packaging/delivery system, packaging component, or packaging material of construction and into an extraction solvent under laboratory conditions.
Leachables – Foreign organic and inorganic chemical entities that are present in a packaged drug product because they have leached into the packaged drug product from a packaging/delivery system, packaging component, or packaging material of construction under normal conditions of storage and use or during accelerated drug product stability studies.
To put it simply, extractables represent a possible impact (worse-case scenario under forced extraction), and studies are performed on the test material (packaging, process component) while leachables refer to the actual impact, where studies are performed on the product in its final packaging (or processing component) under conditions of on-shelf storage.
The leachables study identifies entities that may not all be observed during the preceding extractables study. Thus, the set of leachable entities is not wholly included within the set of extractables, but there is strong overlap between both sets.
Extractables and Leachables Relationships

The responsibility lies with the drug product manufacturer and drug product market authorisation holder to ensure packaging/process components are suitable for use.
Sources of E/L can come from:
- Manufacturing (filters, single-use bags, tubing)
- Primary packaging - External components
- Container-closure system (vials, caps, lids and foils)
- Single-use systems (syringes and intravenous bags)
- Primary packaging - Internal components (gaskets and o-rings, valves, springs)
- Secondary and tertiary packaging (labels and adhesives, inks and colourings)
E/L first came to the attention of the US-FDA in the 1990s, as there was concern over elastomers being used in metered dose inhalers, which could leach polycyclic aromatic hydrocarbons (linked to cancer) into the product. In 2002, there was concern that a possible carcinogen could leach out of IV infusion lines and into the patient. But for many, the defining moment came in 2010 when Johnson & Johnson (J&J)voluntarily recalled products over customer complaints due to a musty odor which was caused by fungicide migrating from wooden shipping pallets through to their secondary and primary packaging systems. It cost J&J an estimated $900 million to manage this issue.
E/L Studies
As per ICH Q9, start with a risk assessment, beginning with what is required for the extractable study and then if a leachable study is required. This can save you money as E/L studies are expensive. The risk assessment (if properly conducted) can help you identify whether you need to do an extractables study on a process component (i.e. it may not come in to contact with the process stream). Biologic drugs pose specific issues due to its complex manufacturing processes, stability issues, large size and extensive surface area’. The extensive surface areas encountered mean that there is a high frequency of potential sites of interaction. Biological drugs also pose analytical challenges in leachables testing due to masking effects/interference, drug dose, mode and frequency of administration.
During the risk assessment, the components of interest will have been identified, which leads into the extractables study. The first step is to define the Analytical Evaluation Threshold (AET), which is derived from the Safety Concern Threshold (SCT); the maximum daily dose of the specific drug product; the duration of use (i.e. one-time administration for acute indications vs a daily administration for chronic indications); and an uncertainty factor to accommodate variations in response to the target compounds. The AET is used to develop the appropriate sample preparation, analysis techniques and serves as a threshold for qualification of potentially harmful compounds.
The purpose of extractable studies is to characterise packaging/delivery systems and components; understand the effects of manufacturing processes (e.g. sterilisation) and their potential leachables; and to allow assessment of patient exposure to chemical entities. Extractable studies are essentially the worst-case scenario where high temperatures and more aggressive solvents are used than in ‘normal’ conditions; there will be a lot more compounds to deal with compared to a leachables study. A couple of key issues to keep in mind with extraction studies:
- It is important to select the right solvent – the solvent could degrade the component
- Ensure all of the component has been tested e.g. if the component is a single use bag, do not cut a sample out of the center of the bag and only test that, there is also tubing, injection ports etc. which can also have an impact
After the extractables study, the data is assessed by a toxicologist to identify the compounds of interest (this can be difficult) and assess the toxicological effect to the patient – not all E/L are toxic. From this conclusion, the leachables study starts. And to throw a spanner in the works, not all leachables may be identified in the extractables study due to issues such as final product-container interaction and degradation of product.
The leachables study is used to assess packaged drug product that has been aged under shelf or simulated conditions for the same classes of compounds as the extraction study. The extraction study builds the worst-case profile, while the leachables assessment focuses on areal-life scenario. Leachables studies should use the same analytical techniques as the extraction study and can be performed concurrently.
For components that have been cleaned/sterilised, the materials must be tested after they have gone through the process - as high autoclave temperatures or irradiation can change the surface of some components.
In certain cases, a leachables study cannot be performed due to a variety of reasons including:
- Drug product formulation may interfere if the study is trying to get to low detection limits
- Required number of samples not available – if it is a novel drug product at early stages of development then there may not be enough to perform all the studies required
- May want to look at the changes in leachables in different batches but variety of batches may not be available – for a typical issue in early-stage development of a biopharmaceutical
- Drug matrix may not be compatible with analytical methods
- Extreme low analytical evaluation threshold needs to be applied due to maximum daily dose, which are analytically not feasible for screening techniques
In these cases, a simulation study may be performed.
E/L studies are typically performed using the following techniques (with validated methods):
- GC-MS (headspace) for volatile organics e.g. adhesives, inks and processing solvents
- GC-MS (Direct injection) for semi-volatile organics e.g. preservatives, PAH’s, plasticisers
- LC-MS for Non-volatile organics e.g. phenol antioxidants
- ICP-MS for Inorganics e.g. elemental Leachables (metals and metal oxides)
A quick note on lyophilised products, even though the product is a powder (low risk) in a glass vial (low risk), this does not mean your product is safe! Firstly, lyophilised products get reconstituted before use, so you will need to consider the aqueous solution and mode of administration (syringe, IV bag etc.). Secondly, there may not be direct contact with the stopper and the lyophilised cake, but there are a lot of volatile and semi-volatile components of the stopper that may be released and ‘fly’ around the vial which can be absorbed into the lyophyilised cake due to it being dry and having a high surface area, These will ‘stick’ to the surface of the lyophilised cake and potentially kickstart unforeseen chemical interactions.
Regulations and Industry Guidance (Pharma/Biologics)
There are quite a few regulations for E/L study requirements, especially from the USP, such as:
- USP <1664> - Assessment of drug product leachables associated with pharmaceutical-packaging delivery systems
- and USP <1665> – Plastic components and systems used in the manufacturing of pharmaceutical products
The EMA also has a few guidelines, including:
- CPMP/QWP/4359/03 and EMEA/CVMP/205/04.5/19/05 on plastic immediate packaging materials
- EMEA/CHMP/QWP/251344/2 - and Limits for genotoxic impurities
The US FDA and regulators in general have to cited companies for not conducting E/L studies. One example from the US FDA being Shilpa Medicare in 2018.


Interestingly the TGA does not state specifically about E/L requirements in the regulations but does mention E/L in a guidance document, 2013 Guidance 18 Impurities in Drug Substances and Drug Products Section 18.3.4. where its begins with “Chemicals leached from glass, plastic, metal or rubber components of medicine containers may cause adverse effects” and then lists what should be done to register a prescription medicine.
There are also quite a few industry groups such as ISO (for medical devices), PDA, BPOG, PQRI and BPSA which have specific guidance documents. BPOG (Biophorum Operations Group) is a group of experts from biopharmaceutical companies around the world who are focused on Single Use Systems and are trying to standardise extractable protocols and to make information more readily available for end users. They have a great risk assessment template for biopharmaceuticals.
However, the one industry group who has really taken on the task of trying to harmonise guidance is the ICH. The ICH have a few guidance documents which bring in to focus issues around E/L such as:
- ICH Q6A Test Procedure and Acceptance Criteria for New Drug Substances and Drug Products and
- ICH M7 Guideline on Mutagenic Impurities,
but there is no international harmonised guidance that exist for E/L assessment and control. ICH released a concept paper in June 2020 for the proposed ICH Q3E Guideline for Extractable and Leachables. This guidance will intend to include chemical and biological products, including drug-device combination products but will leave out medical devices as these are covered in ISO 10993-17: Biological evaluation of medical devices. The new ICH guidance will hope to provide alignment within ICH guidance documents (and hopefully pharmacopeias!); harmonisation of thresholds and safety assessments; control options for risk mitigation; the conduct of E/L studies; the design of an E/L control strategy based on science and risk-based principles; processes to address material and component selection and characterisation; risk assessment, and lifecycle management (including post-approval changes) for container-closure systems, manufacturing systems, and drug delivery device components.
Cell and Gene Therapy (C>) is an interesting area for E/L studies as it a relatively new industry and the focus really is on the manufacturing process (single use systems) rather than the container closure system. Typically, the final product will not be in the final container for very long and as C> is curative the number of doses a patient may receive over their lifetime will be small.
Some issues for leachables studies in C>:
- Major difference between traditional biopharma manufacturing and C> is that in biopharma, the cells are used to make the product, whereas in C> the cells are the product, thus there is no opportunity to separate impurities from the product
- As cells are the product, the cells maybe adherent, thus the product will be ‘sticking’ to the primary packaging, creating a direct source for leachables
The study design comes down to one question – are you looking for a safety or quality outcome? Once this is defined, the usual risk assessment can be performed on the process looking at contact times, manufacturing components, contact area to volume ratio etc.- as with biopharma. But the manufacturing process is different to biopharma, and thus so is the risk assessment e.g. C> processing may have low contact time with components, but they are made in small batches so they have a very high surface area to contact volume ratio (in a few mLs) which is different to a traditional biopharma process which may be dealing with thousands of liters of product.
Currently there is no specific guidelines for E/L studied with C>. The proposed ICH Q3E will hopefully address this. Current guidelines for E/L can be used as a reference but are not really suitable as they use much harsher conditions than you would use for cells e.g. for the choice of solvents, DMSO is readily used in C> but not used routinely as an extraction solvent in E/L. Simulation E/L studies may be preferable for C> due to the small batch size (not enough material) and/or patients may not want to give away a sample of their cells just to do an analytical test rather than creating a therapy.
An understanding of Extractables and Leachables in the manufacturing process and handling of your product is a critical component in assuring patient safety. Regulators are actively assessing companies on their approach with tackling these issues, while international bodies like ICH are working with industry to develop guidelines to assist.
Below is just another example of an observation by the US FDA of a E/L issue, fortunately only a case of a bad odour, with no impact to patient safety.
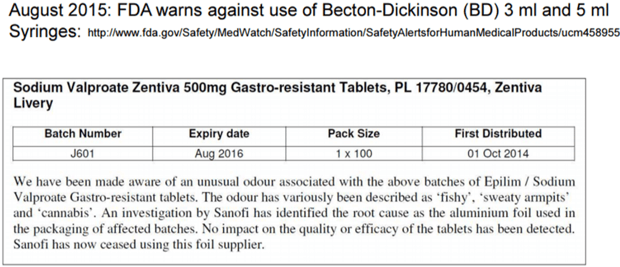
SeerPharma has assisted firms in developing appropriate Extractables and Leachables studies to help assure that appropriate steps are being taken by clients to address these issues.
Contact us if you’d like assistance in the E&L space.