This post was first published in July 2017 and has been updated to reflect changes.
The Therapeutic Goods Administration (TGA) is part of the Australian Federal Government’s Department of Health and is responsible for regulating the supply, import, export, manufacturing and advertising of therapeutic goods, including medicines
Good Manufacturing Practice (GMP) describes a set of principles and procedures that, when followed, helps ensure that therapeutic goods are of high quality. The TGA, as a Pharmaceutical Inspection Convention / Pharmaceutical Inspection Co-operation Scheme (PIC/S) member, has adopted the PIC/S Guide to Good Manufacturing Practice for Medicinal Products.
This article intends to clarify what SeerPharma has found to be unknown or misunderstood in some GMP-licensed organisations relating to the structure and terminology of GMP regulation of medicines in Australia. Understanding the GMP regulatory framework will help pharmaceutical manufacturers to develop a clear and consistent Pharmaceutical Quality System framework.
What is Good Manufacturing Practice (GMP)?
Good Manufacturing Practice (GMP) describes a set of principles and procedures that when followed helps ensure that therapeutic goods are of high quality.
The Hierarchy of GMP Laws: A Snapshot
In Australia, GMP has a legal foundation, consisting of a founding document, The Therapeutic Goods Act, and a set of laws and guidelines. These documents function in a hierarchy (see Figure 1), which determines how they rank in authority. The hierarchical structure is key to determining the purpose of each piece of law within the legal and regulatory framework, and ultimately enforcing their authority and validity.
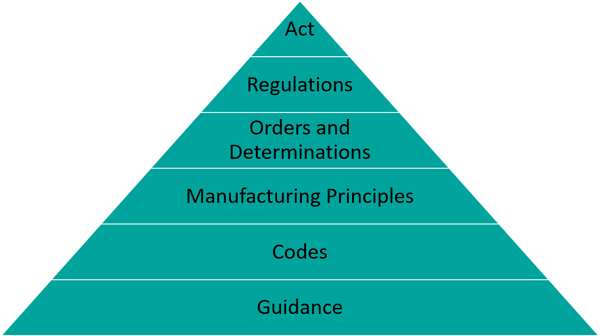
Figure 1: A basic representation of the hierarchy of GMP Laws:
The higher up the hierarchy the document is, the more involved and lengthy the process of amendment is. Broad legal requirements that don’t change frequently are documented in the Act. An example of this is the requirement for manufacturers to hold a valid manufacturing licence. Requirements that may change frequently, like the applicable version of the PIC/S Guide to GMP, are documented further down the hierarchy where amendments are more easily implemented.
The Basics of the GMP Regulatory Framework
Therapeutic Goods Act 1989
The Therapeutic Goods Act 1989 (the Act) sets out the legal requirements for the import, export, manufacture and supply of therapeutic goods in Australia. While specified as 1989, there are periodic amendments to the law, incorporated in “Compilations”.
The Act is supported by the Regulations, and various other Legislative Instruments (including Orders and Determinations) which provide further details of matters covered in the Act.
The Act stipulates that "medicine means:
(a) therapeutic goods (other than biologicals) that are represented to achieve, or are likely to achieve, their principal intended action by pharmacological, chemical, immunological or metabolic means in or on the body of a human; and
(b) any other therapeutic goods declared by the Secretary, for the purpose of the definition of therapeutic device, not to be therapeutic devices."
Therapeutic Goods Regulations 1990
The Therapeutic Goods Regulations 1990 is delegated or subordinate legislation that dictates how the provisions of the Act are applied.
A violation of a regulation can be treated as an offence and enforced as such.
Other Legislative Instruments (examples)
Orders that Goods are Therapeutic Goods / Excluded Goods Orders
Where it is unclear if particular goods are therapeutic, the Therapeutic Goods Administration publishes “Orders that goods are therapeutic goods” or “Excluded goods orders” to define legally what is regulated under the Act.
For example, Orders that Goods are Therapeutic Goods No. 3 of 1999 declared "that products containing shark cartilage, when presented in the form of capsules, pills, tablets or powder, other than as a wholesale product in powdered form for use as an ingredient in food are, for the purposes of the Act, therapeutic goods."
Therapeutic Goods Orders
Therapeutic goods must comply with various standards, which determine the consistency of product quality, including label quality. Many of these standards are international standards. However, sometimes it is necessary to specify an Australian standard, either for a particular type of therapeutic good or to specify particular labelling, packaging or other requirements. In these situations, a “Therapeutic Goods Order” (TGO) is published and some TGOs are accompanied by guidance. Some TGO examples are listed here:
- TGO No. 63 - Standard for Sterile Therapeutic Goods
- TGO No. 89 - Standard for Water for Injections for Parenteral Medicines
- TGO No. 100 - Therapeutic Goods (Microbiological Standards for Medicines) Order 2018
- TGO No. 101 - Therapeutic Goods (Standard for Tablets, Capsules and Pills) Order 2019
Therapeutic Goods Orders are approved by a delegate of the Minister under section 10 (Determination of standards) of the Act, after consultation with the Therapeutic Goods Committee.
Therapeutic Goods Determinations
Determinations define and specify matters relating to the Act. A number of Determinations are published including permissible ingredients (ingredients that are permitted for use in a medicine listed in the Australian Register of Therapeutic Goods), manufacturing principles and things that are / are not biologicals.
Section 36 of the Act allows the Minister for Health to determine manufacturing principles that are to be applied in the manufacture of therapeutic goods. The Therapeutic Goods (Manufacturing Principles) Determination 2020 (Compilation date: 3 June 2024) provides this specification:
Schedule 1 Part 1 - Therapeutic goods other than blood, blood components, haematopoietic progenitor cells etc.
"The manufacture of therapeutic goods to which this Part applies must comply with applicable procedures and requirements in the PIC/S Guide to GMP."
"PIC/S Guide to GMP" is defined as the document titled Guide to Good Manufacturing Practice for Medicinal Products (PE 009-16, 1 February 2022) published by PIC/S...excluding:
^Annex 4 (Manufacture of Veterinary Medicinal Products Other than Immunologicals)
^Annex 5 (Manufacture of Immunological Veterinary Medicinal Products)
^^Annex 14 (Manufacture of Products Derived from Human Blood or Human Plasma)
^Annexes 4 and 5 are excluded because veterinary medicinal products regulation is the responsibility of the Australian Pesticides and Veterinary Medicines Authority (APVMA) – under the Agricultural and Veterinary Chemicals (Administration) Act 1992.
^^Annex 14 is excluded because Australia continues to maintain its own Code of GMP for therapeutic goods that are blood, blood components, haematopoietic progenitor cells and biologicals that do not contain live animal cells, tissues or organs. These products are regulated under Therapeutic Goods (Manufacturing Principles) Determination 2020 Schedule 1 Part 2 which refers to the “Australian Code of Good Manufacturing Practice for human blood and blood components, human tissues and human cellular therapy products (Version 1.0, April 2013) published by the Therapeutic Goods Administration".
Compliance with the PIC/S Guide to GMP is mandatory under the Act and enforcement is generally achieved through conditional licencing. Amongst other conditions of a GMP licence, Section 40 of the Act specifies that the holder of the licence will ensure that:
(i) the goods conform to any standard applicable to the goods; and
(ii) the holder of the licence observes the manufacturing principles in carrying out any steps in the manufacture of the goods under the licence;
Pharmacopoeias
Pharmacopoeias provide standards for pharmaceutical substances and medicinal products. Standards are an important tool in the regulation of the quality of medicines. As from 1 July 2009, the Therapeutic Goods Act 1989 (Chapter 1, section 3) refers to default standards. 'Default standard' means any of the British Pharmacopoeia, European Pharmacopoeia, and United States Pharmacopoeia-National Formulary.
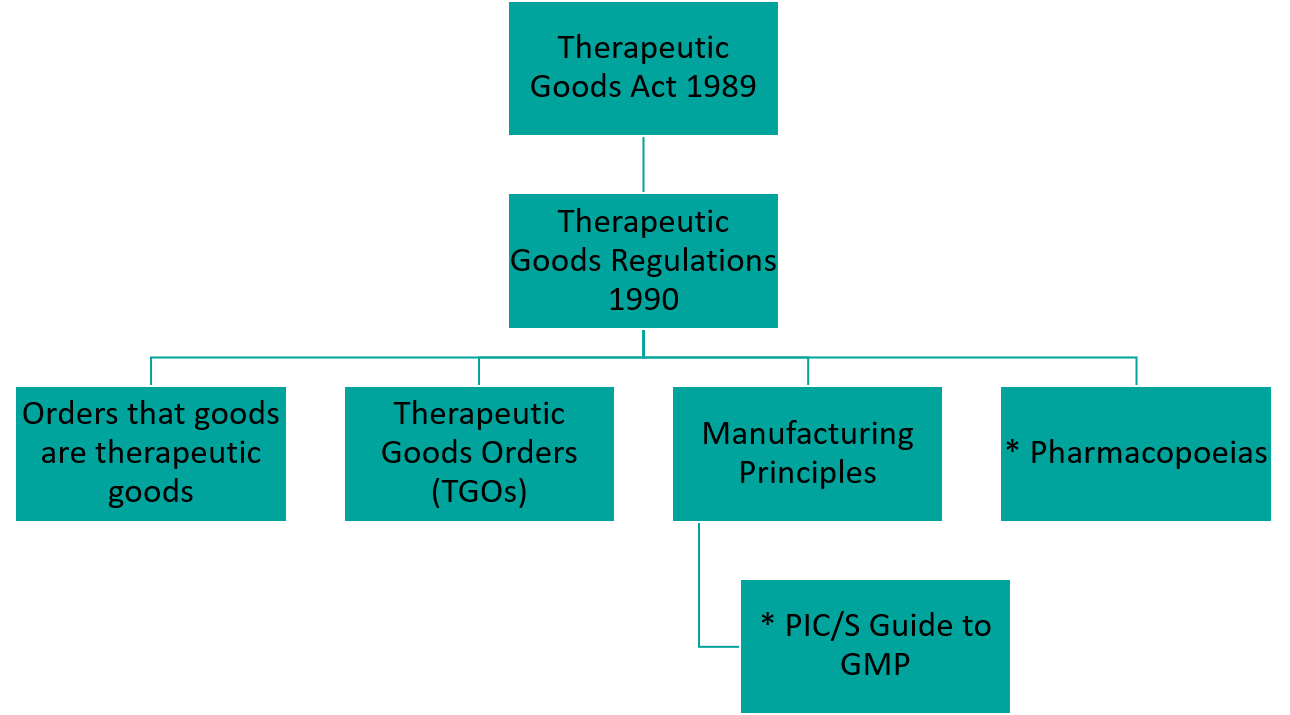
*The PIC/S Guide to GMP and Pharmacopoeias are not published on the Federal Register of Legislation.
Figure 2: A basic representation of the regulatory framework of therapeutic goods subject to PIC/S Guide to GMP compliance. Not all legislative instruments are included.
Guidelines
Guidelines are generally written to ensure that the law is applied consistently and fairly to all parties, but they can often be uncertain in meaning and can result in ambiguity, particularly with regard to enforceability. Strictly speaking, they do not belong as an element of the structure of the hierarchy of laws.
An example of GMP guidance is the Therapeutic Goods Administration's "Release for supply of medicines, Technical guidance on the interpretation of the PIC/S Guide to GMP". The Australian regulator makes it clear that "This guidance is not mandatory or enforceable under law. It is not intended to be restrictive. We recommend following this guidance document to facilitate regulatory obligations being met. The guidance describes a way that a manufacturer may operate to demonstrate compliance with the relevant manufacturing principles (PIC/S Guide to GMP)."
Let's Be Clear: Terminology
GMP terminology can vary between regions and within regions. For example the United States of America’s Food and Drug Administration (FDA) commonly uses “CGMP” as “Current Good Manufacturing Practice”. Within Australia, the term "cGMP" has commonly meant “code of GMP"; which is not specific and could mean the PIC/S Guide to GMP or the Australian Code of Good Manufacturing Practice. Companies must be consistent with the meaning and use of any acronyms and terms to avoid confusion.
In Australia, we must be clear that the “manufacturing principles” is a reference to the full text of the PIC/S Guide to Good Manufacturing Practices for Medicinal Products (excluding annexes 4, 5 and 14) and not just the “Principle” sections that open each chapter.
The manufacturing principles also defines that, "Where the PIC/S Guide to GMP provides that a procedure or requirement ‘should’ be followed, manufacturers of therapeutic goods in Australia must follow that procedure or requirement in order to comply..." However, it also allows manufacturers to deviate from the specified procedure or requirement providing they demonstrate, to the satisfaction of the TGA, that failure to adopt that procedure or requirement or adoption of an alternative procedure does not increase the risks or depart from the record keeping requirements.

Compliance and Enforcement
Although the PIC/S Guide to Good Manufacturing Practices for Medicinal Products is a “guide” by title and is not published on the Federal Register of Legislation, it is a law.
Manufacturers of therapeutic goods for use in humans (other than blood, blood components, haematopoietic progenitor cells etc.) must comply with the PIC/S Guide to GMP as defined by the Act’s reference to “manufacturing principles” and the Therapeutic Goods (Manufacturing Principles) Determination 2020’s specification of the PIC/S Guide to GMP. This legal adoption makes the “guide” a law and we can now see a pathway from the Act (primary legislation) down that gives legal force to the PIC/S Guide to GMP in Australia and justifies the Therapeutic Goods Administration’s inspections of manufacturers in relation to their GMP licence.
The Act defines various levels of criminal and civil offences in relation to breaching a condition of the licence. The worst-case scenario is where the breach “has resulted in, or will result in, harm or injury to any person”, but it is still an offence to breach a condition of the licence irrespective of any outcome or potential outcome.
Ongoing Challenges
We hope that clarifying the GMP regulatory framework will help you develop a clear and consistent Pharmaceutical Quality System framework. We are aware that many companies find this challenging. SeerPharma is proud to have helped many pharmaceutical manufacturers optimise key business and quality system processes for efficiency while maintaining compliance with the PIC/S Guide to GMP.
Contact us if you'd like to explore ways to review or enhance your Pharmaceutical Quality System or address a compliance concern.