In 2014, the FDA issued a guidance document for the Management of Cybersecurity in Medical Devices – Content of Premarket Submissions. Since then, technologies and the interconnectivity of medical devices have evolved rapidly. In response, the FDA released a new draft guidance in April 2022, for comment purposes only - Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions Draft Guidance for Industry and Food and Drug Administration Staff. When final, it will supersede the 2014 version, which is significant due to the recommended changes.
The new paper draws attention to the areas of change since 2014. These are intended to emphasise the importance of ensuring that devices are designed securely are designed to be capable of mitigating emerging cybersecurity risks throughout the device lifecycle and outline the FDA’s recommendations more clearly for premarket submission information to address cybersecurity concerns.

The connectivity of medical device technologies may help improve functionality and benefit, but the connection of medical devices to networks or the internet exposes them to increased cyber threats that can potentially lead to increased risk of harm to patients. These might include:
- denial of intended service or therapy
- alteration of device function to directly cause patient harm
- loss of privacy or alteration of personal health data
Since 2014, significant security events across the healthcare sector have emphasised the importance of cybersecurity to patient safety. We saw the WannaCry ransomware affect hospital systems and medical devices globally. It became apparent that commonly used third-party components within a range of medical devices and clinical services presented vulnerabilities.

These can manifest themselves as impact on direct patient care (medical device) or indirect impacts due to delayed patient care (hospital system). Along with this, the increased connectivity means that individual devices now operate as a single unit of larger medical device systems. Thus, creating a demand for robust cybersecurity control to ensure medical device safety and effectiveness.
The recommendations in the FDA guidance generally align with or expand upon the suggestions in the Pre-Market Considerations for Medical Device Cybersecurity section of the International Medical Device Regulators Forum final guidance “Principles and Practices for Medical Device Cybersecurity,” issued March 2020.
The coverage of the guidance document has increased, and now it also includes the Investigational Device Exemption submissions as well as the ones covered in the previous version. The guidance document has been extended to cover all types of devices under the Federal Food, Drug, and Cosmetic Act, even if they do not require a pre-market submission.
The earlier FDA guidance document required manufacturers to address cybersecurity during the design and development of the medical device (e.g. cybersecurity considerations in design inputs, cybersecurity vulnerability, and management approach as part of design validation). The new approach is to look at the total product lifecycle. The FDA also recommend a cybersecurity framework (developed by NIST) for a company to direct their cybersecurity activities: Identify, Protect, Detect, Respond, and Recover.
For manufacturers, employing processes to "identify, protect, detect, respond and recover" will help keep the cyber security risks of their devices to a minimum. Ultimately, collaboration across all levels of the product's lifecycle will ensure that the benefits of technological advancements in medical devices will outweigh the risks they may present.
In simple terms:
Identify and Protect
Determine the extent to apply security controls based on the device’s intended use and the probable risk to patient harm due to a cybersecurity breach. There must be a balance between safeguards and usability to ensure security controls are appropriate for intended users and situations. For example, controls should not hinder use during an emergency. I draw the analogy here with a GMP manufacturing plant with a control system that has an emergency stop function. You would not want a 2-factor authentication requirement upon pressing the stop button!
Detect, Respond and Recover
- Security issues to be detected, recognised, logged, timed, and acted upon during everyday use
- End users to be informed of appropriate actions to take upon detection of a cybersecurity event
- Ability for retention and recovery of device configuration by an authenticated privileged user
The earlier guidance document talks in high-level terms about the information relating to cybersecurity that manufacturers should be putting in the premarket submission. It also suggests (but doesn’t spell out) that your Quality System Design Control element may need adjusting to effectively manage cybersecurity. The new guidance is more specific about the cybersecurity considerations around 21 CFR 820.30 and ways to satisfy QSR requirements.
The earlier guidance does not talk about transparency of the information about cybersecurity to device users. Still, the latest version puts more emphasis on this to be shared with the users. It is necessary for the user to have information regarding the device’s cybersecurity controls, vulnerabilities, potential risks and other relevant information. The lack thereof can have a detrimental effect on the safety of the medical device.
FDA believes labelling is a great way to transfer the risk information to the user. When the manufacturer submits the premarket submission, it requires them to consider the labelling requirements and how the risk information can be contained in the label. It also becomes an integral part of the QSR in mitigating risks. FDA also recommend that the risk mitigation being transferred to the users be tested during the usability training, so the user is trained on how to mitigate the risk when the situation demands it.
The guidance document also highlights the need for vulnerability management once the device has been handed over to the user. Any upgrade of the software may pose new risks and vulnerabilities that need to be communicated to the user. A proper risk management and corrective and preventative actions strategy must be in place. Thus, when submitting a premarket submission, there is now a need to submit the vulnerability communication plan.
The idea is to analyse the risk of each device when connected to a larger system. Both safety and security risk assessments need to be conducted to fully cover all the possible harm and risks of the device.
A new consideration is adopting a Secure Product Development Framework (SPDF). This framework exists in many device sectors. For the medical device sector, it is a set of processes that reduce the number and severity of vulnerabilities in the medical device throughout the device lifecycle (including development, release, support, and decommission). While the SPDF terminology has not been used in prior FDA guidance, the concepts around secure product development and risk management align with the Quality System Regulation expectations. As cybersecurity continues to evolve, the FDA has said they will continue aligning its terminology to reflect best practices.
Closer to home, the TGA has published two cybersecurity-related guidance documents for the medical device sector – one targeting the industry (manufacturers and sponsors) and one targeting the users.
The industry-related guidance (more aligned with the FDA pre-market guidance) is intended for:
- manufacturers that develop software for use in or as standalone medical devices, such as in Software as a Medical Device (SaMD); this includes devices that incorporate artificial intelligence in their design.
- manufacturers of medical devices (including in-vitro diagnostic medical devices) where devices include components that may be vulnerable to cyber-based threats
- medical device sponsors who are responsible for the supply of medical devices in Australia, to ensure that safety and quality is demonstrated and compliance with the Essential Principles is maintained, e.g.
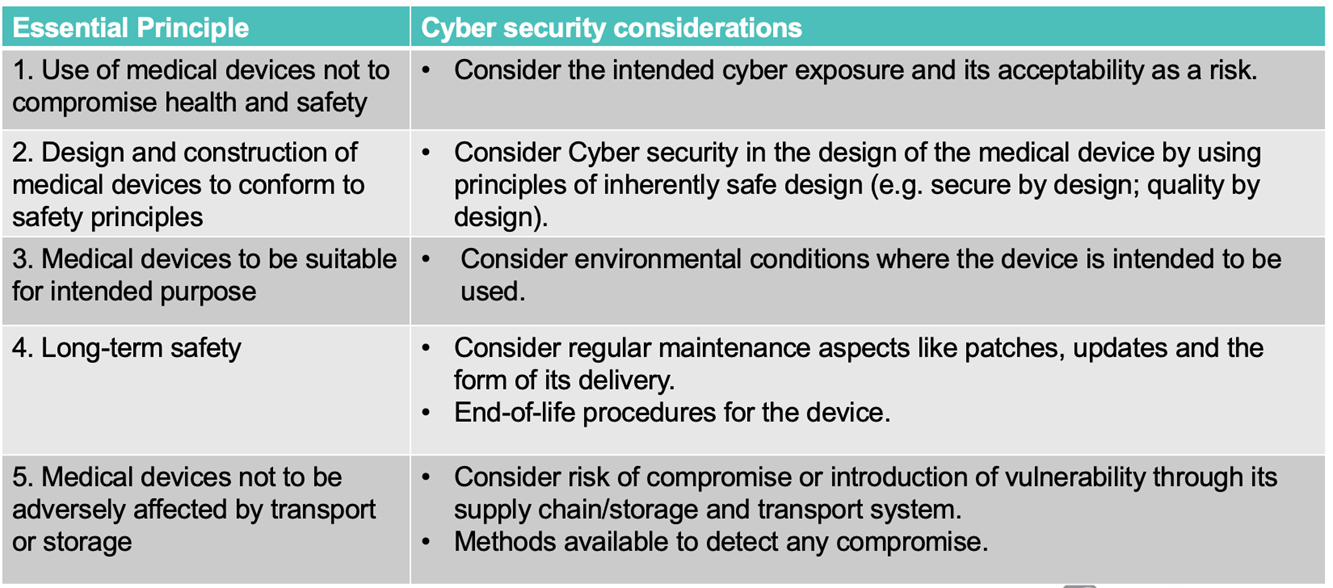
This TGA guidance on cyber security for medical devices is in line with existing regulatory requirements and are guided by and support the Australian Government's cyber security strategy.
The TGA recommends that manufacturers address cyber security risks during the design and development process. This includes:
- general considerations - such as the development approach, administration protocols, application of standards, risk management strategies, infrastructure, manufacturing and supply chain management and provision of information for users
- technical considerations - such as cyber security penetration testing, modularised design architecture, operating platform security, emerging software and Trusted access and content provision
- environmental considerations for the device's intended use - such as connecting to networks and uploading or downloading data
- physical considerations - such as mechanical locks on devices and interfaces, physically securing networks, waste management (preventing the capture of sensitive paper-based information)
- social considerations - such as designing out or minimising social-engineering threats (e.g., phishing, impersonation, baiting, tailgating)
The TGA and FDA recognise that the onus of mitigating and managing cyber security threats is shared across health care facilities, patients, providers, and manufacturers of medical devices.
In particular, medical device manufacturers and health care facilities should take steps to mitigate the risk of cyber security vulnerabilities and maintain proper safeguards throughout the medical device’s lifecycle.

The FDA and TGA are hoping there is a rise in awareness and technology investment for cyber security in the healthcare industry, where historically, the interest of cyber criminals in hacking the healthcare data outweighs the industry’s interest in protecting it.